Compounding
Conditions |
-
Compounded entirely under ISO Class 5 (Class 100) conditions
-
Compounding involves only transfer, measuring, and mixing manipulations with closed or sealed packaging systems that are performed promptly and attentively
-
Manipulations are limited to aseptically opening ampuls, penetrating sterile stoppers on vials with sterile needles and syringes and transferring sterile liquids in sterile syringes to sterile administration devices and packages of other sterile products
|
-
All conditions listed under low-risk level
-
Multiple individual or small doses of sterile products are combined or pooled to prepare a CSP that will be administered either to multiple patients or to one patient on multiple conditions
-
Compounding process includes complex aseptic manipulations other than the single-volume transfer
-
Compounding process requires unusually long duration
-
The sterile CSPs do not contain broad-spectrum bacteriostatic agents, and are administered over several days
|
-
Nonsterile ingredients are incorporated or a nonsterile device is employed before terminal sterilization
-
Sterile ingredients, components, devices and mixtures are exposed to air quality inferior to ISO Class 5 (Class 100)
-
Nonsterile preparations are exposed for not more than 6 hours before being sterilized
-
Nonsterile preparations are terminally sterilized but are not tested for bacterial endotoxins
-
It is assumed that the chemical purity and content strength of ingredients meet their original or compendial specifications in unopened or in opened packages of bulk ingredients
|
| QA Program |
-
-
Describes specific monitoring and evaluation activities
-
Reporting and evaluation of results
-
Identification of follow-up activities when thresholds are exceeded
-
Delineation of individual responsibilities for each aspect of the program
|
See low-risk level. |
See low-risk level. |
| QA Practices |
-
Routine disinfection and quality testing of direct compounding environment
-
Visual confirmation of personnel processes regarding gowning, etc.
-
Review of orders and packages of ingredients to assure correct identity and amounts of ingredients
-
-
Media-fill test procedure performed at least annually for each person
|
See low-risk level. |
See low-risk level. |
| Outcome Monitoring |
Yes |
Yes |
Yes |
| Reports/Documents |
-
Written policies and procedures
-
-
-
Periodic review of quality control documents
|
See low-risk level. |
See low-risk level. |
| Patient and Caregiver Training |
-
Formalized program that includes
-
Understanding of the therapy provided
-
Handling and storage of the CSP
-
Appropriate administration techniques
-
Use and maintenance of any infusion device involved
-
-
|
See low-risk level. |
See low-risk level. |
| Maintaining Product Quality and Control once the CSP leaves the Pharmacy (both institutional based and NICPs) |
-
Packaging, handling, and transport
-
Written policies and procedures including the packaging, handling, and transport of chemotoxic/hazardous CSPs
-
Use and storage
-
Written policies and procedures
-
Administration
-
Written policies and procedures dealing with such issues as handwashing, aseptic technique, site care, etc.
-
Education/Training
-
Written policies and procedures dealing with proper education of patients and caregivers ensuring all of the above
|
See low-risk level. |
See low-risk level. |
| Storage and Beyond-Use Dating |
-
Specific labeling requirements
-
Specific beyond-use dating policies, procedures, and requirements
-
Policies regarding storage
|
See low-risk level. |
See low-risk level. |
| Storage Conditions and Beyond-Use Dating for completed CSP |
In the absence of sterility testing, storage periods (before administration) shall not exceed the following: |
|
Room temperature
2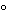 –8 –8
 –20 –20 |
48 hours
14 days
45 days |
Room temperature
2–8
–20 |
30 hours
7 days
45 days |
Room temperature
2–8
–20 |
24 hours
3 days
45 days |
| Finished Product-Release Checks and Tests |
-
Written policies and procedures that address
-
-
Compounding accuracy checks
|
See low-risk level. |
See low-risk level. |
| Finished Product-Release Checks and Tests |
-
Written policies and procedures that address
|
See low-risk level. |
See low-risk level. |
CSP Work
Environment |
-
Appropriate solid surfaces
-
Limited (but necessary) furniture, fixtures, etc.
-
-
|
See low-risk level. |
See low-risk level. |
| Equipment |
-
Written policies and procedures that address calibration, routine maintenance, personnel training
|
See low-risk level. |
See low-risk level. |
| Components |
-
Written policies and procedures that address Sterile components
|
See low-risk level. |
Sterile and nonsterile drug components must meet the compendial standards if available
-
Written policies and procedures that address
|
| Processing: Aseptic Technique |
-
Written policies and procedures that address specific training and performance evaluation
-
Critical operations are carried out in a Direct Compounding Common Area (DCCA)
|
See low-risk level. |
See low-risk level. |
Environmental
Control |
-
Policies and procedures that address
-
Cleaning and sanitizing the workspaces (DCCA)
-
-
Standard operating procedures
|
See low-risk level. |
See low-risk level. |
Verification
Procedures
|
Not required |
Not required |
Yes, recommended |
Verification
Procedures
|
-
Certification of LAFW and barrier isolates every six (6) months
-
Certification of the buffer room/zone and anteroom/zone every six (6) months
-
Bacterial monitoring using an appropriate manner at least monthly
|
See low-risk level. |
See low-risk level. |
Verification
Procedures
-
Personnel Training and Education
|
Initially and annually thereafter
|
See low-risk level. |
See low-risk level. |
;)