Stability Criteria and Beyond-Use Dating
The beyond-use date is the date after which a compounded preparation is not to be used and is determined from the date the preparation is compounded. Because compounded preparations are intended for administration immediately or following short-term storage, their beyond-use dates may be assigned based on criteria different from those applied to assigning expiration dates to manufactured drug products.
Compounders are to consult and apply drug-specific and general stability documentation and literature when available, and are to consider the nature of the drug and its degradation mechanism, the container in which it is packaged, the expected storage conditions, and the intended duration of therapy when assigning a beyond-use date (see Expiration Date and Beyond-Use Date under Labeling in the General Notices and Requirements). Beyond-use dates are to be assigned conservatively. When using manufactured solid dosage forms to prepare a solution or aqueous suspension, the compounder is also to consider factors such as hydrolysis and the freeze-thaw property of the final preparation before assigning a beyond-use date. In assigning a beyond-use date for a compounded drug preparation, in addition to using all available stability information, the compounder is also to use his or her pharmaceutical education and experience.
When a manufactured product is used as the source of active ingredient for a nonsterile compounded preparation, the product expiration date cannot be used to extrapolate directly a beyond-use date for the compounded preparation. However, a compounder may refer to the literature or to the manufacturer for stability information. The compounder may also refer to applicable publications to obtain stability, compatibility, and degradation information on ingredients. All stability data must be carefully interpreted in relation to the actual compounded formulation.
At all steps in the compounding, dispensing, and storage process, the compounder is to observe the compounded drug preparation for signs of instability. For more specific details of some of the common physical signs of deterioration, see
Observing Products for Evidence of Instability under
Stability Considerations in Dispensing Practice  1191
1191
. However, excessive chemical degradation and other drug concentration loss due to reactions may be invisible more often than they are visible.
In the absence of stability information that is applicable to a specific drug and preparation, the following maximum beyond-use dates are recommended for nonsterile compounded drug preparations
1 that are packaged in tight, light-resistant containers and stored at controlled room temperature unless otherwise indicated (see
Preservation, Packaging, Storage, and Labeling in the
General Notices and Requirements).
For Nonaqueous Liquids and Solid Formulations—
Where the Manufactured Drug Product is the Source of Active Ingredient—
The beyond-use date is not later than 25% of the time remaining until the product’s expiration date or 6 months, whichever is earlier.
Where a USP or NF Substance is the Source of Active Ingredient—
The beyond-use date is not later than 6 months.
For Water-Containing Formulations (prepared from ingredients in solid form)—
The beyond-use date is not later than 14 days for liquid preparations when stored at cold temperatures between 2
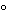
and 8
(36
and 46
F).
For All Other Formulations—
The beyond-use date is not later than the intended duration of therapy or 30 days, whichever is earlier. These beyond-use date limits may be exceeded when there is supporting valid scientific stability information that is directly applicable to the specific preparation (i.e., the same drug concentration range, pH, excipients, vehicle, water content, etc.). See also the beyond-use dating information in the
Labeling section under
Repackaging Into Single-Unit Containers and Unit-Dose Containers for Nonsterile Solid and Liquid Dosage Forms under
Containers 661.