Place the stated volume of the
Dissolution Medium (±1%) in the vessel of the specified apparatus
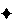
given in the individual monograph,
assemble the apparatus, equilibrate the
Dissolution Medium to 37 ± 0.5
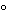
, and remove the thermometer. Place 1 dosage unit in the apparatus, taking care to exclude air bubbles from the surface of the dosage unit, and immediately operate the apparatus at the specified rate
given in the individual monograph
. Within the time interval specified, or at each of the times stated, withdraw a specimen from a zone midway between the surface of the
Dissolution Medium and the top of the rotating basket or blade, not less than 1 cm from the vessel wall.
[NOTE—Where multiple sampling times are specified, replace the aliquots withdrawn for analysis with equal volumes of fresh
Dissolution Medium at 37
or, where it can be shown that replacement of the medium is not necessary, correct for the volume change in the calculation. Keep the vessel covered for the duration of the test, and verify the temperature of the mixture under test at suitable times.
] Perform the analysis
as directed in the individual monograph
using a suitable assay method.
3 Repeat the test with additional dosage form units.
;)
;)
;)
;)
;)
;)