Apparatus—
A liquid chromatograph consists of a reservoir containing the mobile phase, a pump to force the mobile phase through the system at high pressure, an injector to introduce the sample into the mobile phase, a chromatographic column, a detector, and a data collection device such as a computer, integrator, or recorder. Short, small-bore columns containing densely packed particles of stationary phase provide for the rapid exchange of compounds between the mobile and stationary phases. In addition to receiving and reporting detector output, computers are used to control chromatographic settings and operations, thus providing for long periods of unattended operation.
Pumping Systems—HPLC pumping systems deliver metered amounts of mobile phase from the solvent reservoirs to the column through high-pressure tubing and fittings. Modern systems consist of one or more computer-controlled metering pumps that can be programmed to vary the ratio of mobile phase components, as is required for gradient chromatography, or to mix isocratic mobile phases (i.e., mobile phases having a fixed ratio of solvents). However, the proportion of ingredients in premixed isocratic mobile phases can be more accurately controlled than in those delivered by most pumping systems. Operating pressures up to 5000 psi or higher, with delivery rates up to about 10 mL per minute are typical. Pumps used for quantitative analysis should be constructed of materials inert to corrosive mobile phase components and be capable of delivering the mobile phase at a constant rate with minimal fluctuations over extended periods of time.
Injectors—After dissolution in mobile phase or other suitable solution, compounds to be chromatographed are injected into the mobile phase, either manually by syringe or loop injectors, or automatically by autosamplers. The latter consist of a carousel or rack to hold sample vials with tops that have a pierceable septum or stopper and an injection device to transfer sample from the vials to a loop from which it is loaded into the chromatograph. Some autosamplers can be programmed to control sample volume, the number of injections and loop rinse cycles, the interval between injections, and other operating variables.
A syringe can be used for manual injection of samples through a septum when column head pressures are less than 70 atmospheres (about 1000 psi). At higher pressures an injection valve is essential. Some valve systems incorporate a calibrated loop that is filled with test solution for transfer to the column in the mobile phase. In other systems, the test solution is transferred to a cavity by syringe and then switched into the mobile phase.
Columns—For most pharmaceutical analyses, separation is achieved by partition of compounds in the test solution between the mobile and stationary phases. Systems consisting of polar stationary phases and nonpolar mobile phases are described as normal phase, while the opposite arrangement, polar mobile phases and nonpolar stationary phases, is called reverse-phase chromatography. Partition chromatography is almost always used for hydrocarbon-soluble compounds of molecular weight less than 1000. The affinity of a compound for the stationary phase, and thus its retention time on the column, is controlled by making the mobile phase more or less polar. Mobile phase polarity can be varied by the addition of a second, and sometimes a third or even a fourth, component.
Stationary phases for modern, reverse-phase liquid chromatography typically consist of an organic phase chemically bound to silica or other materials. Particles are usually 3 to 10 µm in diameter, but sizes may range up to 50 µm or more for preparative columns. Small particles thinly coated with organic phase provide for low mass transfer resistance and, hence, rapid transfer of compounds between the stationary and mobile phases. Column polarity depends on the polarity of the bound functional groups, which range from relatively nonpolar octadecyl silane to very polar nitrile groups. Liquid, nonbound stationary phases must be largely immiscible in the mobile phase. Even so, it is usually necessary to presaturate the mobile phase with stationary phase to prevent stripping of the stationary phase from the column. Polymeric stationary phases coated on the support are more durable.
Columns used for analytical separations usually have internal diameters of 2 to 5 mm; larger diameter columns are used for preparative chromatography. Columns may be heated to give more efficient separations, but only rarely are they used at temperatures above 60
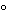
because of potential stationary phase degradation or mobile phase volatility. Unless otherwise specified in the individual monograph, columns are used at ambient temperature.
Ion-exchange chromatography is used to separate water-soluble, ionizable compounds of molecular weight less than 1500. The stationary phases are usually synthetic organic resins; cation-exchange resins contain negatively charged active sites and are used to separate basic substances such as amines, while anion-exchange resins have positively charged active sites for separation of compounds with negatively charged groups, such as phosphate, sulfonate, or carboxylate groups. Water-soluble ionic or ionizable compounds are attracted to the resins, and differences in affinity bring about the chromatographic separation. The pH of the mobile phase, temperature, ion type, ionic concentration, and organic modifiers affect the equilibrium, and these variables can be adjusted to obtain the desired degree of separation.
In size-exclusion chromatography, columns are packed with a porous stationary phase. Molecules of the compounds being chromatographed are filtered according to size. Those too large to enter the pores pass unretained through the column. Smaller molecules enter the pores and are increasingly retained as molecular size decreases. These columns are typically used to measure aggregation and degradation of large molecules (see Size-Exclusion Chromatography section).
Detectors—Many compendial HPLC methods require the use of spectrophotometric detectors. Such a detector consists of a flow-through cell mounted at the end of the column. A beam of UV radiation passes through the flow cell and into the detector. As compounds elute from the column, they pass through the cell and absorb the radiation, resulting in measurable energy level changes.
Fixed, variable, and multi-wavelength detectors are widely available. Fixed wavelength detectors operate at a single wavelength, typically 254 nm, emitted by a low-pressure mercury lamp. Variable wavelength detectors contain a continuous source, such as a deuterium or high-pressure xenon lamp, and a monochromator or an interference filter to generate monochromatic radiation at a wavelength selected by the operator. The wavelength accuracy of a variable-wavelength detector equipped with a monochromator should be checked by the procedure recommended by its manufacturer; if the observed wavelengths differ by more than 3 nm from the correct values, recalibration of the instrument is indicated. Modern variable wavelength detectors can be programmed to change wavelength while an analysis is in progress. Multi-wavelength detectors measure absorbance at two or more wavelengths simultaneously. In diode array multi-wavelength detectors, continuous radiation is passed through the sample cell, then resolved into its constituent wavelengths, which are individually detected by the photodiode array. These detectors acquire absorbance data over the entire UV-visible range, thus providing the analyst with chromatograms at multiple, selectable wavelengths and spectra of the eluting peaks. Diode array detectors usually have lower signal-to-noise ratios than fixed or variable wavelength detectors, and thus are less suitable for analysis of compounds present at low concentrations.
Differential refractometer detectors measure the difference between the refractive index of the mobile phase alone and that of the mobile phase containing chromatographed compounds as it emerges from the column. Refractive index detectors are used to detect non-UV absorbing compounds, but they are less sensitive than UV detectors. They are sensitive to small changes in solvent composition, flow rate, and temperature, so that a reference column may be required to obtain a satisfactory baseline.
Fluorometric detectors are sensitive to compounds that are inherently fluorescent or that can be converted to fluorescent derivatives either by chemical transformation of the compound or by coupling with fluorescent reagents at specific functional groups. If derivatization is required, it can be done prior to chromatographic separation or, alternatively, the reagent can be introduced into the mobile phase just prior to its entering the detector.
Potentiometric, voltametric, or polarographic electrochemical detectors are useful for the quantitation of species that can be oxidized or reduced at a working electrode. These detectors are selective, sensitive, and reliable, but require conducting mobile phases free of dissolved oxygen and reducible metal ions. A pulseless pump must be used, and care must be taken to ensure that the pH, ionic strength, and temperature of the mobile phase remain constant. Working electrodes are prone to contamination by reaction products with consequent variable responses.
Electrochemical detectors with carbon-paste electrodes may be used advantageously to measure nanogram quantities of easily oxidized compounds, notably phenols and catechols.
New detectors continue to be developed in attempts to overcome the deficiencies of those being used.
Data Collection Devices—Modern data stations receive and store detector output and print out chromatograms complete with peak heights, peak areas, sample identification, and method variables. They are also used to program the liquid chromatograph, controlling most variables and providing for long periods of unattended operation.
Data also may be collected on simple recorders for manual measurement or on stand-alone integrators, which range in complexity from those providing a printout of peak areas to those providing chromatograms with peak areas and peak heights calculated and data stored for possible subsequent reprocessing.
Procedure—
The mobile phase composition significantly influences chromatographic performance and the resolution of compounds in the mixture being chromatographed. For accurate quantitative work, high-purity reagents and “HPLC grade” organic solvents must be used. Water of suitable quality should have low conductivity and low UV absorption, appropriate to the intended use.
Reagents used with special types of detectors (e.g., electrochemical, mass spectrometer) may require the establishment of additional tolerances for potential interfering species. Composition has a much greater effect than temperature on the capacity factor, k¢.
In partition chromatography, the partition coefficient, and hence the separation, can be changed by addition of another component to the mobile phase. In ion-exchange chromatography, pH and ionic strength, as well as changes in the composition of the mobile phase, affect capacity factors. The technique of continuously changing the solvent composition during the chromatographic run is called gradient elution or solvent programming. It is sometimes used to chromatograph complex mixtures of components differing greatly in their capacity factors. Detectors that are sensitive to change in solvent composition, such as the differential refractometer, are more difficult to use with the gradient elution technique.
The detector must have a broad linear dynamic range, and compounds to be measured must be resolved from any interfering substances. The linear dynamic range of a compound is the range over which the detector signal response is directly proportional to the amount of the compound. For maximum flexibility in quantitative work, this range should be about three orders of magnitude. HPLC systems are calibrated by plotting peak responses in comparison with known concentrations of a reference standard, using either an external or an internal standardization procedure.
Reliable quantitative results are obtained by external calibration if automatic injectors or autosamplers are used. This method involves direct comparison of the peak responses obtained by separately chromatographing the test and reference standard solutions. If syringe injection, which is irreproducible at the high pressures involved, must be used, better quantitative results are obtained by the internal calibration procedure where a known amount of a noninterfering compound, the internal standard, is added to the test and reference standard solutions, and the ratios of peak responses of drug and internal standard are compared.
Because of normal variations in equipment, supplies, and techniques, a system suitability test is required to ensure that a given operating system may be generally applicable. The main features of system suitability tests are described below.
For information on the interpretation of results, see the section Interpretation of Chromatograms.
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)
;)