General Considerations
The shelf lives of cell and gene therapy products will vary widely, depending on the nature of the product, its intended clinical use, its specific attributes, and the recommended storage, packaging, and shipping conditions. Therefore, it is difficult to draft uniform guidelines regarding stability-study duration and testing frequency applicable to all gene and cell therapy products. In all cases, the stability study should be designed on the basis of scientifically sound principles and approaches and a comprehensive understanding of the final therapeutic product and its intended use. Stability of in-process hold steps, cell and virus banks, critical raw materials, and reference standards also needs to be assessed. A well-designed and executed stability program will provide a high degree of assurance that the product is stable within the specified shelf life.
For viral and nonviral vector gene therapy products and cell therapy products that are not patient-specific, the selection of batches to support license application and final-product labeling should be carried out in accordance with the principles of stability testing, such as those described in ICH guideline Q5C, presented under
Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products  1049
1049
. Stability data should also be collected for bulk material and other in-process points if material is stored before final processing and filling.
Nonviral DNA plasmid vectors are often formulated with specific mixtures of lipids, proteins, or lipoconjugates to form liposomes or encapsulated complexes. Depending on the formulation used, a shelf life of hours to years can be attained. Where a product has a short shelf life, the final formulation may need to be prepared at the clinic just before administration. Instability is frequently observed as aggregation and precipitation. Formation and stability of the formulated complex must be established through validation studies during product development. Stability data should also be collected for major components of the formulated complex, such as the lipids, the liposomes, and the DNA itself.
For many patient-specific cell products including transduced products, each product is unique and often only one lot is prepared for a single patient. In general, the lots tend to be of small volumes, sometimes less than 10 mL, and they may involve products that cannot be frozen and hence have short shelf lives (between 24 and 72 hours) as the cells continue to metabolize their medium. Protocols to establish stability of patient-specific therapy should use materials from multiple donors and at least three lots. Well-characterized banked primary cells may be used in the validation of storage, shipping, and expiration dating if the donors have a range of profiles expected for the patient population to which the therapy will ultimately be directed. The stability of the product under the holding conditions at the medical center should be validated.
Stability-Protocol Development
Formal stability studies to support licensure as well as early phase product stability information gathering should be detailed in a written plan that describes how stability data will be collected and analyzed to support the expiration period of the product. Protocols should follow the format recommended in existing guidelines and include the scope, storage conditions, number of lots to be tested, test schedule, assays to be used, data analysis, and product specifications. Any assay used in a formal stability study for licensure must be validated before the study begins. The specific study design should take into account the reasonably expected possibilities the product may encounter (see Accelerated and Most Appropriate Challenge Conditions) and it should incorporate the latest knowledge in the biological sciences while addressing existing regulatory requirements. For instance, if the final formulation of the product is performed at the clinical site, stability studies on this final formulation should be done to establish the time and conditions under which the product can be held.
Stability studies must verify that the storage conditions maintain the purity and potency of the product, so that the product administered to the patient is still capable of satisfying the stability specifications. These specifications may differ from the release specifications. However, stability specifications must be verified with clinical data. Stability assessment should include assessment of product functionality (potency). The potency assay often has a high degree of inherent variability. Measuring and calculating the decay of product activity by employing the standard statistical methodologies may require multiple, frequent sampling intervals over an extended period of time and may require analysis of more than three production lots to compensate for the variability of the assays. Initial studies to establish a provisional expiration date must be conducted prior to administration to the first patient. Initial studies are also useful for determining which assays are stability-indicating, that is, the best indicators of product degradation. Because existing compendial methods do not address the unique characteristics of cell and gene therapy products, the development of assays that would address these unique characteristics is encouraged.
Accelerated and Most Appropriate Challenge Conditions
The stability-indicating profile of a cell or gene therapy product may vary with time under the influence of a wide variety of environmental conditions, including temperature, extremes in physiological storage conditions, and light. Multifactorial degradation pathways must be considered in the development of a program investigating the effects of these parameters on the stability of the products. Studies should include conditions that are outside of the specified storage ranges, that is, challenge conditions such as those encountered during periods of abnormal storage, shipping, or handling. Examples include brief incubator malfunctions, incubator or cold storage failure, periods of extreme temperature fluctuation due to shipping to hot or cold climates, hypobaric conditions experienced in the cargo hold of a commercial airliner, or temperatures likely to be encountered in the surgical suite. A short exposure to an environmental condition well outside of an established limit and a long exposure to an environmental condition just outside of an established acceptable range may be equally detrimental. The slow and constant rate of product degradation at a specified temperature may increase if a different set of storage conditions is applied. The effect of light on the stability-indicating profile should be investigated if it is scientifically warranted. Special attention should be given to products stored in fluids containing light-sensitive or reactive components that may give rise to cytotoxic by-products.
Studies analogous to accelerated aging studies typically used in pharmaceutical stability-monitoring programs are also useful to determine how the product degrades and which assays are stability-indicating. These studies can be the same as some of those mentioned in the preceding paragraph. Other studies include placing a product at 37
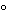
or at 18
, while its normal storage temperature is 25 ± 2
, or placing a lyophilized product in a high-humidity environment. Such studies should be performed before formal stability studies begin, so that the formal studies incorporate the validated stability-indicating assays into the protocol.